
É evidente que no cérebro dos animais, incluindo os humanos, não são as moléculas orgânicas complexas que funcionam diretamente, mas sim estruturas de ordem superior organizadas a partir delas — células nervosas, tecidos e departamentos, mas ainda assim, ainda assim! Em 1994, Leonard Adleman, cientista da computação da Universidade do Sul da Califórnia, em Los Angeles, publicou um artigo científico sobre o “computador sopa de DNA” que ele criou — um tubo de ensaio literalmente preenchido com uma solução aquosa de biomoléculas. A “sopa” revelou-se capaz de resolver o clássico problema do caixeiro-viajante, ou seja, identificar a rota mínima — em termos do tempo gasto em sua passagem — que permitirá contornar um número finito de pontos conectados por um determinado conjunto de caminhos. Para máquinas de von Neumann clássicas, toda a classe desses problemas de otimização combinatória é uma verdadeira dor de cabeça, visto que são NP-completos, ou seja, os matemáticos ainda não descobriram uma maneira de resolvê-los em tempo polinomial. Sim, o experimento de Adleman, com a sessão de cálculo propriamente dita durando cerca de um segundo (e apenas 7 pontos), exigiu uma semana inteira de preparação, mas seu objetivo era praticamente confirmar a própria possibilidade de realizar cálculos de DNA com um nível de paralelismo que máquinas semicondutoras baseadas na lógica de von Neumann nem sequer poderiam sonhar. No entanto, cálculos com base em elementos biomoleculares são um tópico para uma discussão à parte: “computadores” desse tipo sofrem com um nível relativamente alto de erros aleatórios, exigem o desenvolvimento de algoritmos especiais e não são adequados para todos os tipos de tarefas. Mas armazenar dados nas mesmas moléculas de DNA é uma tecnologia que, a julgar por tudo,muito mais próximo da implementação prática.

⇡#Análise do UPS de montagem em rack Ippon Innova RTB 3000

⇡#Senador dos EUA exige esclarecimentos sobre os laços do CEO da Intel com empresas chinesas

⇡#Outra missão da NASA está em risco devido à perda de contato com a sonda que estuda as fendas polares da Terra
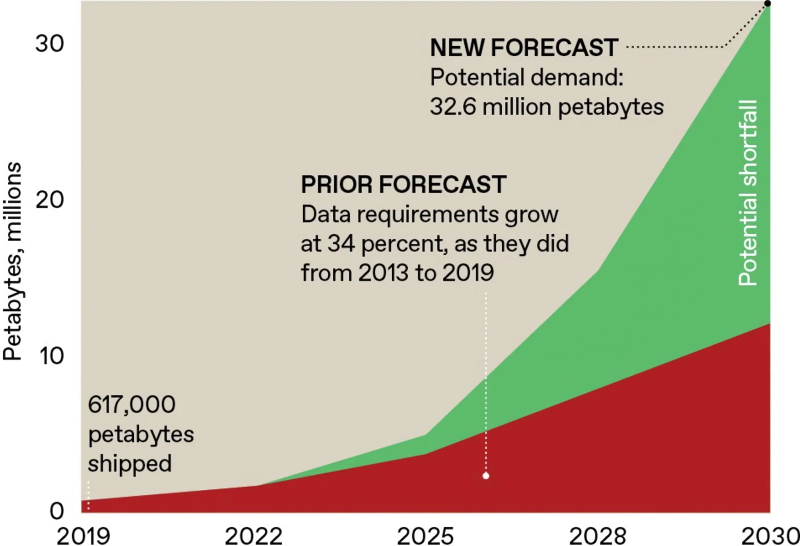
Em 2024, os analistas da Gartner revisaram suas previsões anteriores para o crescimento da demanda global por armazenamento de dados até 2030, em PB (área vermelha), reconhecendo que o desenvolvimento da IA generativa e outras tecnologias digitais adicionarão uma quantidade significativamente significativa de informações (área verde) às estimativas iniciais ano após ano — e até o final de 2025, essa estimativa provavelmente será revisada para cima novamente (fonte: Gartner)
⇡#Tudo já evoluiu antes de nós.
Durante milhares de anos, a humanidade vem descobrindo novas maneiras de registrar dados – em pedra, madeira, tabletes de argila, papiro etc. – até chegar aos dispositivos de armazenamento digital semicondutores e magnéticos. No entanto, a rigor, o que é a própria evolução – portadora do título passageiro da coroa que, a julgar pela presença de uma boa capacidade de pensamento abstrato, pode ser considerada o homem – senão a transformação contínua da informação acumulada pelas gerações anteriores em prol da melhor adaptação (ao seu ambiente) das gerações atuais e, idealmente, das futuras? E como a informação biológica é transmitida entre gerações – para ser repetida e reproduzida em algum lugar, e alterada em algum lugar – ela deve ter portadores materiais. Em meados do século passado, surgiram evidências irrefutáveis de que as moléculas de DNA atuam como tais portadores para praticamente todos os seres vivos no estágio atual de desenvolvimento da vida (no passado, é bem possível que tenha existido um “mundo de RNA” – biopolímeros mais simples, capazes de se autorreproduzir e que agora desempenham principalmente funções de serviço; veja “o dogma central da biologia molecular”). Foi justamente nesses mesmos anos que o famoso Richard P. Feynman, em sua famosa e até profética palestra “Muito espaço no fundo”, sugeriu que o armazenamento natural de informações genéticas pode ser usado para registrar quaisquer dados.
Em princípio — deixando de lado por enquanto a questão da organização técnica dos procedimentos de leitura e escrita — armazenar dados em uma longa cadeia de ácido desoxirribonucleico é extremamente simples. Cada cópia de DNA é uma sequência de nucleotídeos (bases nitrogenadas) de quatro tipos, fixada em uma estrutura de açúcar-fosfato (a mesma estrutura de dupla hélice que todos conhecem das imagens populares dessa molécula): adenina (A), timina (T), guanina (G) e ciazina (C). A estrutura especial de tal molécula — uma dupla hélice, cujos nucleotídeos de ambos os ramos são pareados por ligações de hidrogênio — garante sua mais alta estabilidade: a uma temperatura de +9 °C, o DNA individual — fora de uma célula viva — é preservado por pelo menos 2 mil anos, e a estimativa da preservação de tal molécula a -18 °C já é de 2 milhões de anos, o que certamente excede a vida útil garantida de qualquer suporte de dados magnético ou semicondutor.

À esquerda, há um esboço de uma seção de uma molécula de DNA com seus principais componentes indicados; no centro, há um diagrama explicando a correspondência mútua dos nucleotídeos na cadeia (em frente a A, em uma molécula intacta, apenas T pode ser localizado; em frente a C, apenas G); à direita, está a fórmula estrutural de uma seção de DNA com bases nitrogenadas e uma estrutura de açúcar-fosfato (fonte: Human Origin Project)
E embora uma única molécula seja literalmente um objeto nanométrico (o diâmetro da dupla hélice é de 2 nm, a distância entre pares adjacentes de nucleotídeos ao longo das cadeias de açúcar e fosfato é de 0,34 nm), de um ponto de vista puramente técnico, ela é excelente para armazenamento de dados bastante confiável. É claro – foi criada dessa forma pela evolução biológica que durou bilhões de anos! Por exemplo, a estrutura de dupla fita do DNA não apenas simplifica a formação de um par de cópias idênticas da informação genética armazenada nessa molécula (com foco na reprodução), mas também protege essa informação de danos, por exemplo, causados por fótons ultravioleta de alta energia, ou, mais precisamente, permite que mecanismos intracelulares especialmente desenvolvidos detectem áreas danificadas em uma das cadeias, as cortem e, em seguida, construam o fragmento necessário na área liberada ao longo da cadeia complementar de nucleotídeos da segunda cadeia.
É claro que, como portadoras de informação, as moléculas de DNA provavelmente serão armazenadas e usadas para leitura e escrita separadamente, fora das paredes celulares – na forma de uma solução aquosa, por exemplo, como no experimento de Adleman –, mas terão que ser trabalhadas usando métodos químicos; semelhantes aos que operam em células vivas. Felizmente, esses métodos são essencialmente insubstituíveis: se as ligações de hidrogênio entre nucleotídeos em fitas complementares são relativamente frágeis – elas são destruídas pelo aquecimento mesmo antes da fervura, por exemplo – então as bases nitrogenadas dentro de cada fita estão ligadas tão fortemente que podem ser separadas por um ácido muito forte ou pelo uso de enzimas especiais, nucleases quiméricas, especialmente projetadas para tais operações. Grosso modo, uma nuclease é um composto proteico composto por duas unidades estruturais, capaz de primeiro se ligar seletivamente a certas sequências de nucleotídeos no DNA alvo e, em seguida, catalisar sua clivagem (“cortar a molécula”) neste local. No início da década de 2010, um método ainda mais eficaz de engenharia genética foi proposto, o CRISPR/Cas, onde moléculas curtas de RNA, em vez de proteínas, são usadas para reconhecer o sítio alvo, mas o próprio princípio de “detecção e corte” continua a depender de mecanismos biológicos.

Princípio de organização de armazenamento de dados de longo prazo em DNA (fonte: IEEE Spectrum)
Atualmente, as tecnologias de sequenciamento de DNA, ou seja, a leitura da sequência de nucleotídeos “gravada” nele, bem como sua síntese – o problema inverso de organizar bases nitrogenadas díspares, que podem ser consideradas unidades elementares de dados codificados, em uma molécula forte e durável – foram muito bem desenvolvidas, literalmente em escala industrial. A etapa-chave é o registro de dados: a informação destinada ao armazenamento em DNA é traduzida do código binário para uma sequência de nucleotídeos designada (onde A, na primeira aproximação, pode codificar “00”, T – “01”, etc.), e então esses nucleotídeos são costurados da maneira adequada. Tecnologicamente, o processo de costura lembra mais a superposição sequencial de moléculas umas sobre as outras no processo de síntese química em vários estágios, e inicialmente poderia levar meses para várias dezenas de nucleotídeos, mas o trabalho está sendo feito constantemente para acelerá-lo e simplificá-lo.
Em 2016, pesquisadores da Universidade de Washington, juntamente com colegas da Microsoft, relataram ter conseguido codificar 35 arquivos diferentes, variando em tamanho de 29 KB a 44 MB, com um volume total de mais de 200 MB, usando aproximadamente 13 milhões de oligonucleotídeos – fragmentos curtos de DNA de 200 bases nitrogenadas ou menos. E não apenas codificar, mas oferecer um método para busca subsequente e leitura seletiva de dados registrados em nível molecular. Essa conquista é notável precisamente porque, embora anteriormente fosse possível em laboratórios codificar informações em DNA montado artificialmente e extraí-las, pela primeira vez na prática foi demonstrado um método comparativamente rápido e preciso para organizar o acesso aleatório a um volume tão impressionante de dados armazenados em nível molecular.

Em 2018, pesquisadores revelaram um protótipo de desktop de uma máquina universal para escrever, armazenar e ler dados em moléculas de DNA (fonte: Microsoft)
⇡#Muitos dados
Por que é tão importante poder acessar mídias de armazenamento molecular à vontade para selecionar os arquivos necessários, já que, por exemplo, fitas magnéticas, ainda usadas para arquivamento, são geralmente aceitas como dispositivos de acesso sequencial? Se calcularmos formalmente quantos nucleotídeos individuais caberão em 1 mm³ em condições normais (com alguma tolerância para que não estejam localizados próximos uns dos outros, mas formando duplas hélices clássicas), obtemos um limite teórico superior muito encorajador para a densidade de gravação de informações no DNA – cerca de 1 Ebyte para este volume, ou, em termos de massa, 215 Pbytes em cada grama (assumindo, no entanto, codificação de dois bits sem redundância – o que, como veremos mais tarde, seria excessivamente otimista). Portanto, se nos limitarmos a simplesmente decodificar todas as informações registradas nessas macromoléculas de uma só vez – e tecnicamente isso é o mais fácil de fazer, porque, como já foi dito, o sequenciamento de DNA é hoje relativamente rápido e barato – teremos que registrar as informações decifradas em algum meio externo em relação ao tubo de ensaio com os “dados dissolvidos” e, em seguida, procurar os arquivos necessários lá. Tal abordagem não faz sentido no próprio uso de macromoléculas como dispositivos de armazenamento independentes, cuja principal vantagem é sua densidade de armazenamento incomparável. Também não devemos esquecer que tanto a síntese de DNA quanto seu sequenciamento são operações com um nível extremamente alto (para os padrões dos computadores semicondutores de von Neumann) de erros e, portanto, é necessário prever alguns mecanismos para suacompensação/correções, pelo menos somas de verificação banais e/ou duplicação (ou melhor ainda, alinhamento) de sequências de dados.
Um método proposto por pesquisadores em 2016 contornou essa limitação de forma eficaz, embora talvez não totalmente elegante, utilizando primers oligonucleotídicos (fragmentos muito curtos – cerca de 20 bases nitrogenadas – de uma única fita de DNA) em combinação com a reação em cadeia da polimerase (PCR). Essa reação, agora amplamente utilizada em pesquisas médicas, permite criar – com esforço relativamente modesto – cópias de um determinado fragmento de DNA disponível. A história da PCR remonta a 1957; o importante neste caso é que seu uso aumenta a concentração de uma sequência nucleotídica específica – se ela estiver inicialmente presente na amostra – em várias ordens de magnitude, tornando a detecção da sequência desejada praticamente trivial. De fato, é precisamente graças à PCR que algumas doenças infecciosas são diagnosticadas hoje de forma tão rápida e confiável (com base nas “impressões digitais moleculares” do material genético dos microrganismos que as provocam), a pertença de materiais biológicos encontrados na cena do crime a um suspeito é confirmada/refutada, etc.
Essencialmente, a inovação dos pesquisadores da Universidade de Washington e da Microsoft foi que eles desenvolveram uma extensa biblioteca de primers-alvo na etapa anterior à codificação dos dados registrados no DNA, que foram então anexados a ambas as extremidades de cada uma das sequências de nucleotídeos contendo um determinado fragmento de informação. O grande DNA final foi então formado a partir de fragmentos marcados dispostos ponta a ponta, e na etapa de busca, o acesso aleatório foi implementado de forma extremamente natural: se for conhecido quais cadeias de bases nitrogenadas são marcadas com determinados primers, a reação de PCR torna possível aumentar a concentração dessas cadeias na solução a um nível tal que o sequenciamento subsequente seja realizado de forma bastante rápida e com o mínimo de erros (para cuja correção, por sua vez, os pesquisadores desenvolveram algoritmos adequados para esse método).

Um exemplo de codificação de uma sequência de caracteres de texto (neste caso, “DN referencial”) em código binário, depois em uma sequência de nucleotídeos, seguido pela síntese de moléculas de DNA, seu sequenciamento e decodificação – a saída é a mesma sequência de letras novamente (fonte: Wyss Institute)
Como, de fato, é realizada a codificação primária de dados moleculares — a criação de fragmentos sintéticos básicos, que são então formatados nas extremidades por primers catalogados e usados para formar o DNA total final? Aqui, a química molecular vem ao resgate: nucleotídeos individuais são ligados entre si, formando uma cadeia polimérica, por meio de ligações fosfodiéster sequenciais, ao longo de uma série de etapas. Primeiro, dois monômeros (nucleotídeos individuais; digamos, A e G) são ligados — em um volume macroscópico, é claro; durante uma reação química com ativação do grupo fosfato, e não com pinças convencionais sob um microscópio igualmente convencional — então o próximo nucleotídeo necessário (digamos, C) é adicionado à solução do dímero resultante, obtendo-se assim um trímero, e assim por diante — até que toda a cadeia com a configuração necessária seja formada. No processo, é necessário monitorar cuidadosamente a homogeneidade dos produtos resultantes, introduzindo reagentes protetores para bloquear reações indesejadas. Atualmente, métodos mais avançados (rápidos, baratos e que proporcionam um alto nível de pureza do produto em cada etapa) utilizando a síntese de fosforamidita em fase sólida em sintetizadores automáticos são conhecidos, de modo que a criação de sequências curtas de nucleotídeos não pode mais ser considerada um problema sério. Em 2018, empresas como a GenScript Biotech e a Integrated DNA Technologies ofereciam a qualquer pessoa que desejasse o serviço de sintetizar fragmentos de DNA de fita dupla com até várias centenas de pares de bases por apenas 10 a 40 centavos por par; hoje, o preço é consideravelmente mais baixo.
Claro, não é tão simples assim: mesmo considerando a altíssima qualidade de cada síntese de amidofosfito separadamente – em 99% dos casos, o nucleotídeo necessário é adicionado à cadeia de nucleotídeos que está sendo formada na etapa seguinte –, como resultado do procedimento, que requer a implementação sucessiva de centenas dessas reações, apenas 37% do produto final atende exatamente às necessidades do cliente. De fato, esta é uma das razões mais importantes pelas quais o método descrito – justificando-se plenamente para aplicações comerciais, automatizado com segurança – só consegue produzir oligonucleotídeos, ou seja, moléculas relativamente curtas (em comparação com o DNA natural): a natureza quase exponencial do acúmulo de erros torna a síntese de longas cadeias de bases sem sentido. No entanto, a montagem de moléculas longas a partir de oligonucleotídeos marcados com primers cuidadosamente selecionados é bastante justificada – porém, aqui é necessário envolver não tanto competências químicas, mas sim matemáticas e cibernéticas.

Os primers colocados nas extremidades da cadeia de nucleotídeos alvo permitem que a reação em cadeia da polimerase detecte de forma confiável as regiões de DNA necessárias e as corte para sequenciamento posterior (fonte: IEEE Spectrum)
⇡#Os assuntos de dias muito futuros
Para começar, é necessário proteger ao máximo as informações armazenadas no DNA contra o surgimento e o acúmulo de erros que inevitavelmente surgirão durante reações químicas que ocorrem em volumes pequenos (até microlitros), mas ainda macroscópicos – em comparação com uma única molécula. No início da década de 2010, dois grupos de pesquisadores, de Harvard e do Instituto Britânico de Bioinformática Europeia, propuseram simultaneamente um esquema bastante complexo, mas, segundo suas garantias, extremamente confiável, para a codificação de informações para dispositivos de armazenamento de DNA. Em primeiro lugar, recomendou-se a transição da representação binária de dados para um sistema ternário (com uma base não de “0” e “1”, mas de “0”, “1” e “2”). Por que ternário? Sim, porque longas sequências de nucleotídeos idênticos (AAAAAA, GGGG, até mesmo o CC banal) são frequentemente reproduzidas com erros durante o sequenciamento e, portanto, faz sentido garantir uma alternância de bases em sua cadeia o mais frequente possível desde o estágio inicial – idealmente sem sequer permitir repetições simples. Por esta razão, não há correspondência biunívoca entre os “0”, “1” e “2” do sistema de codificação ternário e os quatro tipos de bases – A, T, G, C – do portador de DNA no esquema proposto. Em vez disso, uma regra mais complexa está em vigor, permitindo uma algoritmização clara: “se dois símbolos idênticos seguem em sequência na codificação baseada na base 3, diferentes nucleotídeos são usados para designá-los, com um certo deslocamento”. Exemplo: seja uma determinada letra na codificação ternária representada como 20112, então na tradução em nucleotídeos ela corresponderá à sequência TAGAT – aqui, a primeira de um par de unidades adjacentes é representada como G, e a segundajá como A, embora literalmente agora o mesmo A denotasse “0”. Acontece que é perfeitamente possível compilar uma tabela de correspondências biunívocas de tais substituições precisamente no caso de codificação baseada na base 3 usando 4 “letras” de nucleotídeos — e então usar essa tabela tanto ao registrar informações no DNA quanto na fase de leitura.
O segundo ponto importante: os dados no DNA são gravados com uma redundância incrível, para os padrões de mídia semicondutora ou magnética: 75%. Ou seja, se a macromolécula final longa for composta por blocos elementares de 100 nucleotídeos (em certo sentido, estes são análogos de setores na superfície de um disco rígido magnético), então, em cada bloco subsequente, apenas os últimos 25 pares de bases serão originais; os primeiros 75 repetirão exatamente os 75 pares finais do bloco anterior. A razão é óbvia: embora portas lógicas moleculares para operações com oligonucleotídeos já tenham sido propostas, sua adição a um sistema destinado apenas ao armazenamento de dados complicará desproporcionalmente e o tornará mais caro. E a falta de lógica computacional impossibilita o uso de um dos meios mais eficazes de detectar/corrigir erros: gerar e verificar regularmente somas de verificação. Assim, a mera redundância da representação ternária-quaternária de dados em DNA (um sistema ternário de representação convencional de símbolos; quatro nucleotídeos para sua materialização física) permanece, em tal situação, a única garantia confiável contra a ocorrência de um número significativo de erros tanto durante o registro quanto na leitura. É evidente que o armazenamento de informações sobre macromoléculas não se torna mais barato: em 2013, quando pesquisadores americanos e britânicos propuseram o método descrito, o custo médio de gravação de 1 MB de informação em DNA (com sua divisão em blocos curtos, com a síntese sequencial dos oligonucleotídeos correspondentes em laboratórios especializados e sua subsequente união em uma única molécula) atingiu 12 mil dólares; a leitura do mesmo 1 MB – 220dólares. Hoje, esses valores são ordens de magnitude menores, e é justamente por essa razão que o DNA, como portador de longo prazo de dados arquivados, vem atraindo cada vez mais atenção prática.

Uma maneira de ser confiável – no sentido de maximizar a proteção contra erros, cuja probabilidade de ocorrer para cada molécula individual durante uma reação química, onde muitas delas estão envolvidas, é codificar uma sequência de texto em uma sequência de nucleotídeos (fonte: Instituto Europeu de Bioinformática)
No entanto, como frequentemente acontece tanto na microeletrônica quanto na química molecular, todo tipo de sutileza começa mais adiante (mas são elas que tornam as pesquisas científicas e de engenharia verdadeiramente empolgantes!). Parece que aqui está, um esquema simples e bem-sucedido para organizar dados em DNA: assim como uma trilha de gravação em um disco magnético é marcada em setores, uma macromolécula longa e portadora de informações é composta de blocos de oligonucleotídeos, cada um dos quais marcado com primers que a identificam de forma única. Para extrair os dados, resta apenas verificar o catálogo de primers (claro, localizado em um meio externo, provavelmente um clássico – semicondutor ou magnético), preparar as polimerases correspondentes, realizar a PCR e obter na saída a seção do DNA original na qual a informação necessária está registrada.
Infelizmente, na prática, a implementação desse esquema bastante transparente está associada a dificuldades consideráveis: os primers devem ser inicialmente selecionados de modo que se liguem apenas aos oligonucleotídeos correspondentes, e não a quaisquer outros que formem o DNA final – e quanto mais longo for o processo, mais trabalhosa e não rotineira essa tarefa se torna. Além disso, existem pequenas diferenças entre os nucleotídeos que formam o DNA: formalmente, o mesmo tipo de ligações de hidrogênio entre os pares que compõem os “degraus” da conhecida escada em espiral é ligeiramente mais forte no caso da conexão G-C do que para A-T. Isso, por sua vez, significa que, se um determinado primer contiver muitas bases A e T, sua conexão com a seção correspondente do DNA será, ao contrário, mais fraca do que a de outros, ou seja, as operações com ele durante a PCR serão realizadas com um tempo mais comprimido, o que quase certamente levará ao aparecimento de erros adicionais durante o sequenciamento.
Além disso, diferentes sequenciadores (dispositivos automatizados para, na verdade, sequenciar genomas) de diferentes desenvolvedores são únicos no sentido de que tendem a cometer certos erros com mais frequência: alguns às vezes (e quanto maior a molécula, mais frequentemente) substituem bases – em vez de A, colocam, digamos, C nesta posição; outros acidentalmente “perdem” – não incluem na decodificação final do DNA – blocos inteiros de oligonucleotídeos; em outros, pares individuais desaparecem sem deixar vestígios da cadeia de bases durante o processo de decodificação, etc. Felizmente, sabendo a que tipo de erros um determinado sequenciador específico está sujeito, é possível, puramente matematicamente, fornecer mecanismos adicionais para detectar e compensar seus erros futuros no nível da codificação ternário-quaternária de dados. Mas isso, por sua vez, priva o DNA de sua universalidade como portador de dados! Acontece que, para decodificar informações armazenadas com a expectativa de reprodução em uma instalação específica, será necessário usar apenas essa mesma instalação; e mesmo substituí-lo por um sucessor direto – no qual alguns erros específicos se manifestarão com menos frequência – pode simplesmente piorar, e não melhorar, a qualidade da reprodução dos dados. Surge a pergunta: vale a pena fazer tanto alarde sobre o armazenamento de DNA, esperando transmitir informações em macromoléculas ao longo de séculos e milênios, se não há sequer uma garantia firme de que nossos descendentes distantes (ou alguns reptilianos visitantes de Nibiru) serão capazes de descobrir, em princípio, onde exatamente esses brincalhões pré-históricos colocaram dados significativos nessa cadeia específica de nucleotídeos – e de que maneira engenhosa eles os codificaram?

Alguns sequenciadores utilizam filtros nanopore para ler o DNA original, através do qual essas longas moléculas passam como espaguete pelos buracos de uma peneira, permitindo que uma base nitrogenada após a outra seja identificada sequencialmente. No entanto, como o tempo alocado para cada identificação é tão curto (caso contrário, mesmo o menor DNA, com milhões de nucleotídeos, levaria um tempo excessivamente longo para ser processado), erros são inevitáveis: cada uma das quatro bases possíveis tem alguma chance de ser lida como uma das outras três, com probabilidades ligeiramente diferentes para diferentes nucleotídeos (fonte: IEEE Spectrum).
Em última análise, verifica-se que a criação de dispositivos de armazenamento de DNA requer a cooperação mais estreita de especialistas nas áreas de biologia sintética e criptografia (ou, mais precisamente, teoria da codificação de dados, uma área verdadeiramente vasta da matemática aplicada) para eliminar da forma mais eficaz os problemas causados pela imperfeição das atuais tecnologias de síntese orgânica e sequenciamento. No entanto, um resultado semelhante pode aparentemente ser alcançado desenvolvendo essas mesmas tecnologias de maneiras não triviais – com a transição, em particular, da síntese/sequenciamento “úmido” tradicional em um volume macroscópico de líquido para a construção de estruturas poliméricas ordenadas, nas estruturas fibrosas das quais as moléculas individuais de DNA são tecidas. Aqui, em vez de se preocupar com a PCR, pesquisadores da Universidade da Carolina do Norte e da Universidade Johns Hopkins propuseram, em 2024, retornar ao método natural de leitura, cópia e reescrita de DNA, ou seja, usando moléculas de RNA especializadas para esses fins (o que, é claro, dá origem a uma série de outros problemas, mas remove uma quantidade considerável dos atualmente relevantes). Outro método potencialmente inovador foi desenvolvido nos últimos anos na China: ele envolve o uso não dos próprios nucleotídeos que formam os genes como portadores diretos de dados, mas de “epibites”, que são formados com base nos nucleotídeos no processo de metilação (que, aliás, também desempenha um papel significativo no campo natural e biológico do funcionamento do DNA – veja “epigenética”). Como neste caso, em princípio, nada precisa ser recortado e colado – a própria cadeia dupla de bases permanece inalterada –Os processos de leitura e escrita são significativamente acelerados, atingindo impressionantes 40 bits/s para armazenamento de dados biomoleculares (o protótipo da Universidade de Washington e da Microsoft de 2018, que mencionamos, processou 5 bytes por 21 horas).
Finalmente, já em 2025, pesquisadores da Universidade do Texas em Austin tiveram a ideia de sintetizar moléculas semelhantes a DNA a partir de “polímeros de sequência definida”. A vantagem indiscutível desse método é a capacidade de não se limitar ao nível mais básico de codificação: em vez das quatro “letras naturais” G, C, A e T, os texanos criaram imediatamente 256 componentes elementares com diferentes propriedades eletroquímicas, o que tornou possível aumentar significativamente a precisão de sua identificação em solução — e, consequentemente, acelerar as operações de leitura/gravação, reduzindo simultaneamente a frequência de erros. E até mesmo o fato de o polímero sintético experimental ser destruído durante o processo de leitura pode ser usado a favor — usando essa abordagem, por exemplo, para trocar dados sensíveis com a necessidade de garantir que eles não tenham sido comprometidos durante a transmissão.
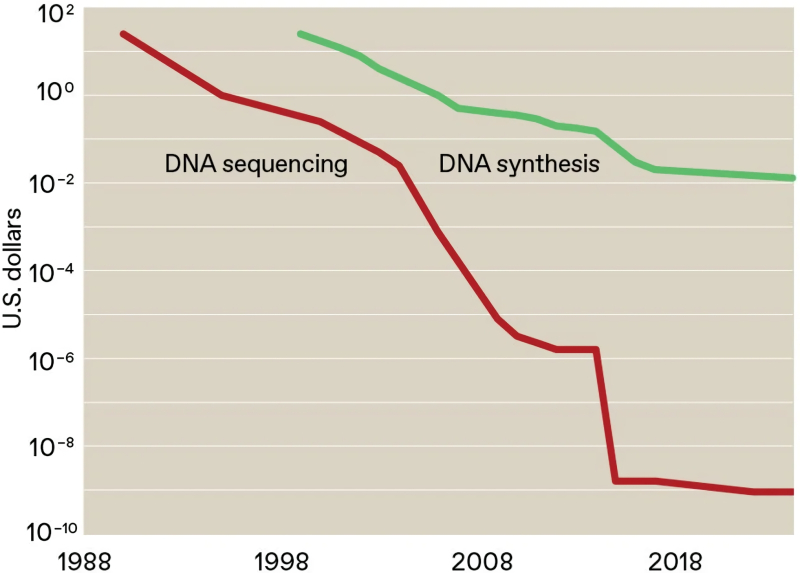
Ao longo de 35 anos, o custo do sequenciamento laboratorial de um único par de bases caiu de US$ 25 para menos de um milionésimo de dólar, mas a síntese continua muito cara para uma implementação verdadeiramente em grande escala de dispositivos de armazenamento de DNA (fonte: IEEE Spectrum)
Em suma, por enquanto, o principal impulsionador do progresso no campo dos dispositivos de armazenamento de DNA continua sendo a incrível densidade de armazenamento que eles fornecem — pelos padrões de quase todos os outros suportes de dados: todos os 120 ZB, que no início de 2024 foi estimado como o volume de informações disponíveis pela Internet, deveriam teoricamente caber em apenas 1 cm3 dessas moléculas (não em uma solução, é claro, mas simplesmente compactadas umas às outras — e se deixarmos de lado a questão de quanto tempo levará para preparar os dados para codificação e a formação real desse cubo). Ao contrário das fitas magnéticas, o DNA, sob condições de armazenamento devidamente organizadas, não se degrada por pelo menos séculos e não requer eletricidade para manter seu estado ou testes periódicos da operabilidade do suporte com possível reescrita subsequente — para arquivar enormes quantidades de dados (sim, estamos olhando para você, treinando bancos de dados para treinar IA cada vez mais em larga escala), esta é quase uma opção ideal.
Em 2020, diversas universidades de pesquisa e laboratórios comerciais e financiados pelo orçamento fundaram a DNA Data Storage Alliance — um consórcio internacional que visa acelerar a superação das barreiras que atualmente impedem o desenvolvimento e a implementação do armazenamento de DNA. E esta é, antes de tudo, uma velocidade inaceitavelmente baixa (do ponto de vista das tarefas cotidianas de arquivamento), que, para chegar aos 2 Gbit/s, alcançáveis pelas fitas magnéticas modernas, terá que ser de alguma forma aumentada dos baixos valores atuais de cerca de 300 mil nucleotídeos por segundo; nucleotídeos; há muito menos dados “líquidos” úteis devido à enorme redundância da gravação — pelo menos quatro casas decimais. O objetivo, desnecessário dizer, é ambicioso — vamos ver o quão rápido os desenvolvedores se moverão em direção a ele! No mínimo, a necessidade de enormes instalações de armazenamento de dados com capacidade de energia mínima, ou melhor ainda, zero, está se tornando mais urgente a cada ano, o que significa que há um incentivo considerável para aprimorar os dispositivos de armazenamento de DNA.
Materiais relacionados
- Cientistas tiveram a ideia de armazenar dados em um análogo plástico do DNA – ele será denso e confiável.
- Os chineses propuseram gravar dados em diamantes para sempre – a densidade será 10.000 vezes maior do que em um DVD.
- Uma tecnologia foi desenvolvida para registrar dados no DNA existente.
- Cientistas criaram a base para futuros computadores de DNA que armazenam e processam dados simultaneamente.
- Cientistas encapsularam DNA em âmbar artificial, criando uma solução de armazenamento de dados superdensa e durável.
- Cientistas chineses apresentaram uma base para a criação de computadores universais baseados em DNA.
- Compatibilidade biológica.